Over 80% of clinical trials fail to meet recruitment timelines, often due to non-compliance with Good Clinical Practice (GCP) in researcher guidelines.
In today’s complex clinical research landscape, obtaining ICH E6(R2,R3) certification is paramount for ensuring compliance with international standards and significantly improving study success rates. This certification not only enhances the quality and integrity of clinical trials but also plays a crucial role in safeguarding the rights, safety, and well-being of trial participants.
This blog will guide you through the comprehensive process of achieving ICH E6(R2,R3) certification, providing you with essential knowledge to navigate the intricacies of Good Clinical Practice and elevate your research standards. Whether you’re an experienced professional or new to the field, this information will prove invaluable in advancing your career in clinical research and maintaining rigorous regulatory compliance.
Key takeaways:
- ICH E6(R2,R3) fundamentals: Grasp the guideline’s core principles and its critical importance in clinical research.
- Training and education: Explore comprehensive ICH E6(R2,R3) training programmes and resources for certification.
- Compliance strategy: Learn to develop and implement a robust plan tailored to your organisation’s needs.
- Documentation best practices: Master the art of proper record-keeping for ICH E6(R2,R3) compliance.
- Audit readiness: Discover effective strategies for conducting internal audits and preparing for external inspections.
- Certification process: Navigate the journey of selecting a certification body and applying successfully.
- Continuous improvement: Implement ongoing compliance efforts and stay updated with the latest regulatory changes.
What is ICH E6(R2,R3) certification?
ICH E6(R2,R3) certification demonstrates compliance with the Good Clinical Practice (GCP) guidelines established by the International Council for Harmonisation. This internationally recognised standard ensures clinical trials are conducted ethically and scientifically, prioritising participant safety and data credibility.
In clinical research, ICH E6(R2,R3) certification is crucial. It provides a unified standard for the European Union, Japan, and the United States, facilitating mutual acceptance of clinical data by regulatory authorities. This certification showcases commitment to the highest quality and ethical standards in clinical trials.
The ICH E6(R2,R3) history dates back to 1996 when the original guideline was first published. Since then, it has evolved to address the increasing complexity of modern clinical trials. The lateset R3 revision focuses on:
- Subject protection and data integrity
- Risk-based quality management
- Enhanced sponsor oversight
- Computerized systems and electronic records
One significant update in ICH E6(R3) is the emphasis on Risk-Based Monitoring. This approach allows for a more flexible and targeted monitoring strategy based on the specific risk level associated with a trial, improving efficiency without compromising quality.
Key benefits of obtaining ICH E6(R2,R3) certification include:
- Enhanced credibility in the global research community
- Improved efficiency in conducting multi-regional clinical trials
- Better protection of trial subjects’ rights and safety
- Increased likelihood of regulatory acceptance of trial data
- Overall improved quality of clinical research
Bottom line: by aligning with the World Medical Association’s Declaration of Helsinki, thereby maximising subject rights, safety, privacy, and well-being. This certification is an essential benchmark for quality and integrity in clinical research, fostering trust among stakeholders and advancing global health initiatives.
We offer comprehensive Good Clinical Practice (GCP) course to help you achieve certification and excel in your clinical research endeavours.
Step 1: Understand the ICH E6(R2,R3) Guideline
Understanding the ICH E6(R2,R3) guideline is crucial for obtaining certification. Let’s break down the key aspects:
Overview of the ICH E6(R2,R3) document structure:
The guideline is organised into sections covering GCP principles, stakeholder responsibilities, and essential trial documents. It provides a comprehensive framework for ensuring ethical conduct and scientific integrity in clinical research.
Key changes and additions in the R2 revision:
The R2 revision, adopted in 2016, introduced significant updates:
- Enhanced sponsor responsibilities
- Clarified monitoring processes
- Stronger emphasis on risk management
Key Changes and Additions in the R3 Revision (2025):
The R3 revision introduces several important updates and enhancements to strengthen the Good Clinical Practice (GCP) framework:
- Expanded sponsor and investigator responsibilities to reflect advances in technology and trial methodologies.
Focus on risk-based approaches and quality management:
A central theme is the implementation of Risk-Based Quality Management (RBQM). This approach encourages sponsors and investigators to identify, assess, and mitigate risks throughout the clinical trial process, ensuring more efficient and effective trial management.
Resources for accessing and studying the guideline:
Access the full ICH E6(R2,R3) guideline on the official ICH website. Regulatory bodies like the FDA and EMA provide additional guidance documents and training materials to aid in understanding and implementation.
Tips for effective comprehension and interpretation:
- Start with an overview, then delve into specific sections
- Participate in focused training workshops or webinars
- Join professional forums to discuss interpretations with peers
- Regularly review updates and addendums to stay current
Bottom line: A thorough understanding of the ICH E6(R2,R3) guideline is the foundation for developing compliant clinical trial practices and ensuring the protection of human subjects while maintaining data integrity. It’s essential for successful certification and implementation.
Step 2: Complete ICH E6(R2,R3) Training and Education
To obtain ICH E6(R2,R3) certification, comprehensive training and education are essential. Let’s explore your options for mastering this crucial guideline:
Training Programs:
Introductory courses for newcomers to clinical research
Advanced workshops for experienced professionals
Specialised programmes focusing on specific ICH E6(R2,R3) aspects
Certification preparation courses
Reputable Providers:
Organisations like the MRCT Center offer excellent ICH E6(R2,R3) courses. We at Whitehall Training also provide comprehensive ICH E6(R2,R3) training tailored to your needs.
Training Formats:
Online courses: Offer flexibility and self-paced learning
In-person training: Provides hands-on experience and networking opportunities
Hybrid programmes: Combine both online and in-person elements
Key Topics Covered:
Ethical considerations in clinical research
Quality management systems
Risk-based approaches to clinical trials
Essential documents and record-keeping
Sponsor and investigator responsibilities
Safety reporting and monitoring
Continuous Learning:
Stay updated on regulatory changes, industry best practices, emerging technologies in clinical research, and real-world applications of ICH E6(R2,R3) through ongoing education.
Remember, quality training ensures proper interpretation and application of ICH E6(R2,R3) principles in clinical research practices. By investing in your education, you’ll be well-equipped to implement the guideline effectively and maintain compliance in your clinical trials.
Bottom line: Comprehensive training is crucial for successful ICH E6(R2,R3) certification and implementation. Choose a programme that fits your needs and commit to ongoing learning to stay at the forefront of good clinical practice.
Step 3: Develop a Compliance Strategy
Developing a robust compliance strategy is crucial for successful ICH E6(R2,R3) implementation and certification. Let’s break down the key components:
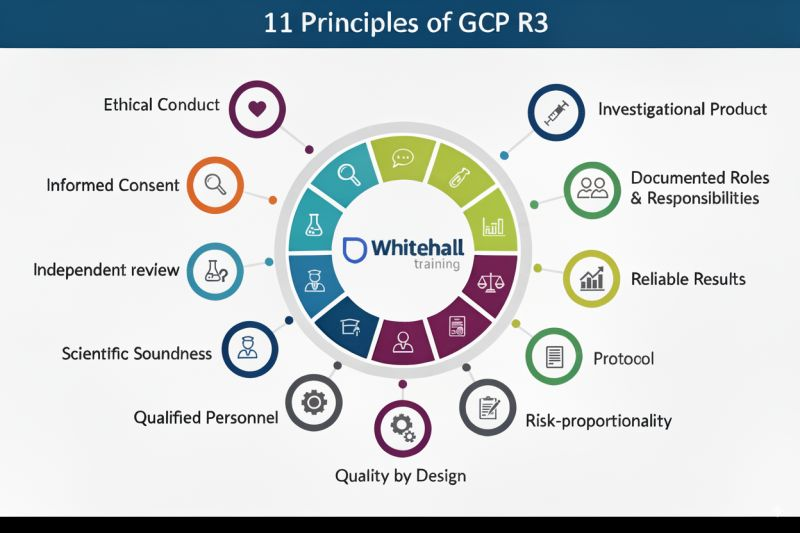
- Implement the 11 principles of good clinical practice:Ensure your team understands and applies these principles daily – Cover ethical conduct, protocol adherence, and data integrity
- Create a robust Quality Management System:Focus on consistently meeting customer requirements and enhancing satisfaction – Encompass all aspects of clinical research operations, from study design to reporting
- Develop an effective monitoring plan:Oversee trial progress, ensure protocol compliance, and protect participants’ rights – Consider risk-based monitoring approaches to optimize resource allocation
- Establish a comprehensive risk management plan:Identify potential risks to trial integrity and participant safety – Assess likelihood and impact, outlining mitigation strategies – Regularly review and update throughout the trial lifecycle
- Integrate ICH E6(R2,R3) principles into existing processes:Update standard operating procedures, training programs, and documentation practices – Ensure all team members understand how principles apply to their specific roles
We offer Good Clinical Practice (GCP) course to help you develop a robust compliance strategy. Our expert-led training ensures you’re well-equipped to meet guideline requirements and demonstrate your commitment to high-quality clinical research practices.
Bottom line: A well-structured compliance strategy is essential for successful ICH E6(R2) implementation and certification. By addressing these components thoroughly, you’ll be well-positioned to meet the guideline’s requirements and showcase your dedication to excellence in clinical research.
Step 4: Document Compliance and Maintain Records
Thorough documentation is the backbone of ICH E6(R2,R3) compliance. It’s not just about keeping records; it’s about creating a clear, traceable history of your clinical trial processes. Let’s explore the key aspects of documentation and record-keeping for your certification journey.
Key areas requiring documentation include:
- Informed consent processes
- Adverse event reporting
- Protocol deviations
- Data collection and management
- Investigator and staff qualifications
- Equipment calibration and maintenance
Best practices for maintaining accurate and complete records:
- Implement standardised documentation processes
- Train staff on proper documentation techniques
- Regularly review and audit records
- Use clear, concise language
To organise and retrieve records efficiently, consider implementing an electronic document management system with features like version control, audit trails, and secure access.
Data integrity and traceability are crucial. Ensure all processes and decisions can be traced back to their origins, creating a clear audit trail. The investigator should ensure the accuracy, completeness, legibility, and timeliness of the data reported to the sponsor in the CRFs and all required reports. Any changes or corrections should be dated, initialled, and explained to maintain data integrity.
Remember to maintain investigator records for at least 2 years after the last approval of a marketing application in an ICH region, or until there are no pending or contemplated marketing applications, or at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product.
Accurate record keeping is emphasised in ICH GCP E6(R2,R3), which stresses the importance of maintaining complete records that are attributable, legible, contemporaneous, original, accurate, and complete.
We understand the challenges of documentation in clinical trials. Our ICH E6(R2,R3) training courses can help you master these essential skills and ensure your compliance with the latest guidelines.
Bottom line: Proper documentation is crucial for demonstrating compliance and passing audits or inspections. By implementing robust documentation practices, you’ll be well-positioned to achieve and maintain ICH E6(R2,R3) certification.
Step 5: Conduct Internal Audits and Prepare for Inspections
Regular internal audits are a cornerstone of maintaining ICH E6(R2,R3) compliance and preparing for certification. These audits provide invaluable Internal Audit Benefits, offering tailored insights to identify and rectify process flaws while proactively enhancing operational efficiency. By conducting thorough self-assessments, organisations can ensure they’re meeting the stringent requirements of ICH E6(R2,R3) and be better prepared for external inspections.
To maximise the effectiveness of your internal audit process, develop a comprehensive audit plan and schedule. This should outline the frequency of audits, areas to be covered, and responsible personnel. Aim to audit all critical aspects of your clinical trial processes at least annually, with more frequent checks for high-risk areas.
When conducting internal audits, focus on key areas that align with ICH E6(R2,R3) principles:
- Quality management systems
- Risk-based monitoring processes
- Data integrity and management
- Investigator and site management
- Informed consent procedures
- Safety reporting and pharmacovigilance
- Protocol compliance
- Documentation and record-keeping
As you identify non-conformities during audits, address them promptly and implement corrective actions. This proactive approach not only ensures ongoing compliance but also demonstrates a commitment to continuous improvement – a key aspect of ICH E6(R2).
Preparing for regulatory inspections is the final crucial step. Use the insights gained from internal audits to refine your processes and documentation. Conduct mock inspections to familiarise your team with the inspection process and identify any remaining gaps in your compliance efforts.
We offer comprehensive ICH E6(R2,R3) training courses to help you master these audit and inspection preparation techniques. Our expert-led programmes ensure you’re well-equipped to maintain compliance and achieve certification.
Bottom line: Regular audits and inspection readiness are essential for maintaining ICH E6(R2,R3) compliance and increasing your chances of successful certification. By implementing a robust internal audit program and preparing diligently for inspections, you’ll be well-positioned to demonstrate your commitment to quality and compliance in clinical research.
Step 6: Choose a Certification Body and Apply for Certification
Selecting the right certification body is crucial for obtaining ICH E6(R2,R3) certification. When researching accredited bodies, look for organisations with a strong reputation in clinical research and recognition by regulatory authorities.
Consider these factors when choosing:
- Track record of successful certifications
- Expertise in Good Clinical Practice (GCP)
- Support and guidance throughout the process
- Costs and timelines
- Accreditation status and industry recognition
The application process typically involves submitting a formal request with detailed information about your organisation, clinical trial processes, and quality management system. Be prepared to demonstrate how you’ve implemented the 11 principles of good clinical practice outlined in the ICH E6(R2,R3) guideline.
Required documentation and evidence may include:
- Standard Operating Procedures (SOPs)
- Training records
- Quality management system documentation
- Clinical trial protocols and reports
- Evidence of risk management practices
- Proof of ethical conduct and subject wellbeing measures
- Data integrity safeguards
Timeframes can vary, generally taking several months from application to certification. This process may include initial assessment, documentation review, on-site audits, and corrective actions if necessary. Costs also vary, so request detailed quotes from multiple bodies to ensure you’re getting the best value for your investment.
We offer comprehensive Good Clinical Practice (GCP) course to help prepare you for certification. Our expert-led programmes cover all aspects of GCP, ensuring you’re well-equipped for the application process and ongoing compliance.
Remember, proper preparation is key to successful certification. It demonstrates your commitment to ICH E6(R2) Compliance and ethical and scientific quality standards in clinical trials, enhancing your credibility in the field.
Bottom line: Selecting the right certification body and thoroughly preparing for the application process is crucial for obtaining ICH E6(R2,R3) certification. This step ensures that your organisation meets international standards for clinical trials, protecting subject rights and data credibility while enhancing your competitiveness in the industry.
Step 7: Implement Continuous Improvement Processes
Obtaining ICH E6(R2,R3) certification is not a one-time achievement but an ongoing commitment to excellence in clinical research. The ICH E6(R2) Compliance guideline emphasises the importance of ongoing compliance and improvement to ensure the safety of research participants and the integrity of data. This final step in your certification journey focuses on implementing processes that foster continuous improvement and maintain long-term compliance.
To stay updated on ICH E6(R2,R3) developments:
- Subscribe to regulatory authority newsletters
- Join professional associations
- Attend relevant conferences and webinars
- Engage with industry peers through forums and networking events
These resources provide valuable insights into upcoming changes and best practices in the field.
Implementing feedback mechanisms and performance metrics is crucial for identifying areas of improvement:
- Establish a system for collecting and analysing feedback from staff, participants, and stakeholders
- Use key performance indicators (KPIs) to measure the effectiveness of your clinical trial processes
- Implement digital tools to streamline compliance processes and improve data management
Regular review and update of clinical trial processes is essential to ensure ongoing compliance:
- Schedule periodic assessments of your procedures, documentation, and training programs
- Conduct internal audits to identify potential gaps or inefficiencies
- Stay proactive in addressing issues before they become significant problems
Fostering a culture of quality and compliance is perhaps the most critical aspect of continuous improvement:
- Encourage open communication about potential issues
- Reward staff for identifying and addressing problems
- Provide ongoing training to keep everyone informed about the latest ICH E6(R2,R3)requirements and best practices
- Promote a mindset of continuous learning and adaptation
Bottom line: Continuous improvement ensures long-term compliance and maintains the value of ICH E6(R2,R3) certification. By implementing these strategies, you not only safeguard your certification but also contribute to the advancement of ethical and efficient clinical research practices, ultimately leading to safer and more effective clinical trials.
Summary: Achieve and Maintain ICH E6(R2) Certification for Research Excellence
Obtaining ICH E6(R2,R3)Certification is a crucial step for clinical research professionals, ensuring compliance with international ethical and scientific quality standards. Let’s recap the key steps to achieve this certification:
- Thoroughly understand the ICH E6(R2,R3) guideline
- Complete comprehensive training and education
- Develop a robust compliance strategy
- Implement thorough documentation practices
- Conduct regular internal audits and prepare for inspections
- Choose a reputable certification body and apply
- Establish continuous improvement processes
Remember, compliance with ICH E6(R2,R3) is an ongoing journey. Regularly review and update your practices to maintain certification and stay aligned with evolving industry standards.
The benefits of ICH E6(R2,R3) certification are substantial:
- Enhanced credibility in the research community
- Improved data quality and integrity
- Increased efficiency in clinical trial processes
- Demonstration of commitment to ethical research practices and patient safety
If you haven’t started your certification journey yet, now is the time to begin. The path to excellence in clinical research starts with a strong foundation in Good Clinical Practice. Good Clinical Practice (GCP) course provides comprehensive training to guide you through the process.
For further learning and support, consider exploring these valuable resources:
- Regulatory body websites (e.g., FDA, EMA)
- Professional associations in clinical research
- Specialized training providers
To conclude, ICH E6(R2) is pivotal in advancing clinical research quality and integrity. By pursuing this certification, you’re not just fulfilling a requirement – you’re contributing to the advancement of medical science and the protection of human subjects. Find out how to get your Good Clinical Practice (GCP) certification today.
Bibliography
![How to Get an ICH E6(R2,R3) Certification [2025]](https://wp.whitehalltraining.com/wp-content/uploads/2025/11/68b6be6eb3569e524962630f_good-clinical-practice-11-scaled.jpg)




![Good Clinical Data Management Practice Guidelines [2025]](https://wp.whitehalltraining.com/wp-content/uploads/2025/11/68b6be6eb3569e5249626330_research-ethics_29-scaled.jpg)

