Discover how AI, data, and innovation are modernising clinical research — improving speed, compliance, and patient outcomes.
🎥 Watch now: https://youtu.be/qrb1VDpblLw?si=ovQkAuPAoKFGz1NF

Discover how AI, data, and innovation are modernising clinical research — improving speed, compliance, and patient outcomes.
🎥 Watch now: https://youtu.be/qrb1VDpblLw?si=ovQkAuPAoKFGz1NF
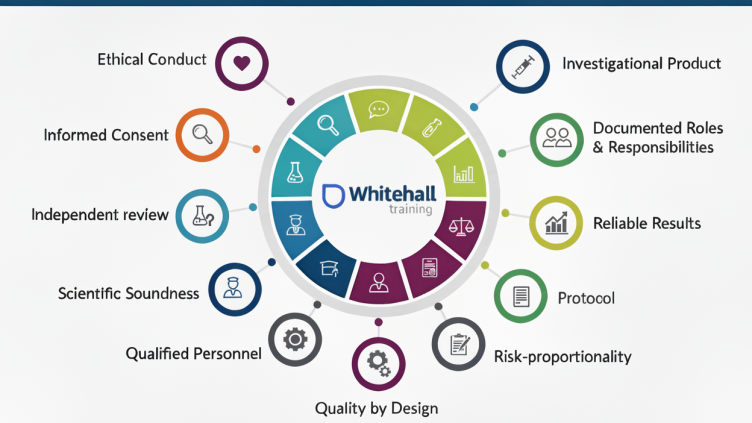
The world of clinical research is evolving — and so are the standards that govern it.
If you’re working in clinical trials, regulatory affairs, or quality assurance, understanding the ICH E6 Good Clinical Practice (GCP) guidelines is essential.
With the R3 revision emphasizing risk-based quality management, digital data integrity, and decentralized trials, now is the perfect time to upskill and stay compliant.
Here’s how you can get certified in ICH E6 (R2/R3) in 2025:
1️⃣ Choose a recognized training provider — Look for accredited institutions (e.g., TransCelerate, DIA, Coursera, or accredited GCP academies).
2️⃣ Complete the ICH E6 course modules — Covering R2 fundamentals and R3 updates (risk management, sponsor responsibilities, data integrity).
3️⃣ Pass the final assessment — Most programs include a quiz or case-based evaluation to test your GCP understanding.
4️⃣ Earn your certificate — Valid proof of GCP compliance and competency.
5️⃣ Keep it current — With R3 still in adoption, continuous learning is key.
Pro tip: If you’re already certified in R2, an R3 bridging course can help you transition smoothly to the updated standard.
Staying ahead in GCP means embracing change — and R3 is setting the stage for smarter, more transparent, and technology-driven clinical trials.

In the fast-paced world of clinical research, “Good Clinical Practice” (GCP) is not just a guideline—it is the global standard that safeguards ethical conduct and scientific integrity. For years, ICH GCP E6 (R2) has been the playbook for industry professionals. However, with the arrival of ICH GCP E6 (R3), the landscape is shifting significantly.
The transition from R2 to R3 is more than just a version update; it is a modernization effort designed to address the realities of 21st-century clinical trials. From decentralized trials to digital health technologies, understanding these changes is critical for sponsors, investigators, and clinical teams aiming to stay compliant and audit-ready.
While R2 introduced the concept of risk-based monitoring, R3 takes it a step further, embedding “Quality by Design” (QbD) into the very DNA of the protocol. Here are the primary differences that every clinical professional needs to know:
One of the most visible changes is in terminology. R3 retires the term “subject” in favor of “participant.” This is not merely semantic; it reflects a fundamental shift toward patient centricity. The new guidelines emphasize that trials should be designed with the participant’s burden in mind, ensuring their rights, safety, and well-being are prioritized not just in theory, but in the practical design of the study.
When R2 was written, the idea of a fully remote trial was a novelty. R3 explicitly addresses the use of computerized systems and digital health technologies. It provides a framework for decentralized clinical trials (DCTs), validating remote monitoring, e-consent, and wearable data collection as standard, acceptable practices—provided they are validated and secure.
R2 encouraged risk-based thinking, but R3 mandates a risk-proportionate approach. This means that quality management should be proactive rather than reactive. Instead of fixing errors after they happen, trial designs must anticipate risks to critical data and processes. “Critical to Quality” factors must be identified before the first participant is even recruited.
With the explosion of data sources (e.g., electronic health records, apps, wearables), R3 places a heavier burden on Data Governance. Sponsors and investigators must ensure that external data systems are reliable and that the flow of data—from a patient’s device to the final study report—remains traceable and secure.
It is a common misconception that you can wait until a guideline is fully enforced globally before adapting. However, major regulatory bodies like the EMA and FDA are already aligning their expectations with the principles of R3.
Why does this matter?
The most common pitfall during a regulatory transition is assuming that experienced staff “already know GCP.”
The shift to R3 requires active re-training. It is no longer sufficient for staff to hold a certificate from five years ago. Clinical Research Associates (CRAs), Project Managers, and Investigators need to understand how to apply risk-proportionate strategies and how to manage digital data streams.
Key Training Focus Areas for R3:
The transition to ICH GCP E6 (R3) represents a smarter, more flexible, and more ethical future for clinical trials. By embracing these changes now—through updated SOPs and comprehensive training—organizations can ensure they are not just compliant, but are leading the way in modern clinical research.

1.0 Introduction: The Hidden World of Medical Research
When we picture a clinical trial investigator, the image is often of a dedicated scientist in a lab coat, meticulously collecting data, analyzing results, and pursuing the next medical breakthrough. They are the face of scientific progress, focused on the rigorous details of research. While this picture is not wrong, it barely scratches the surface of their true role.
Behind the scenes, the investigator’s responsibilities extend far beyond the scientific method. They are a complex blend of operational manager, legal guardian, and ethical steward. Their work is governed by a strict set of international standards known as Good Clinical Practice (GCP)—a framework born from the darkest moments of medical history, like the post-WWII Nuremberg Code, to ensure that research is always conducted ethically and that the rights, safety, and well-being of trial participants are paramount.
This article pulls back the curtain on three of the most surprising and impactful responsibilities that define an investigator’s work. These duties reveal a role that is less about pure science and more about a profound commitment to the human beings at the heart of medical discovery.
Beyond designing experiments and interpreting data, a clinical trial investigator is the operational leader responsible for all site and participant-related matters. They function as the chief executive of their research site, ensuring that the entire trial is not just scientifically sound but also logistically robust and ethically managed from start to finish.
This managerial role encompasses a wide range of duties that are critical to the trial’s success and the safety of its participants. The investigator is ultimately accountable for:
This CEO-like function is the bedrock of a trial’s integrity. A scientific protocol, no matter how brilliant, is worthless if the site is understaffed, the product is stored incorrectly, or the team is improperly trained. The investigator’s managerial skill is what translates a scientific plan into a viable, ethical, and trustworthy human endeavor.
A common misconception is that informed consent is merely the act of a participant signing a form. In reality, it is a deep and continuous ethical process—a conversation that must never be rushed. The fundamental goal is not to get a signature, but to ensure the participant truly understands every aspect of the trial so their decision to join is completely voluntary and well-informed.
This responsibility becomes even more profound when dealing with participants who may be vulnerable. Investigators must follow specific, and often surprising, procedures to safeguard their autonomy:
Furthermore, consent is not a static event. If any significant new information about the trial’s risks or benefits emerges, the investigator must re-inform participants and obtain their re-consent to continue. This ongoing dialogue underscores the deep ethical commitment to ensuring participation remains voluntary and fully understood at every stage, safeguarding the rights of every individual. This constant attention to consent reinforces the most fundamental principle of medical ethics: a person’s autonomy is inviolable. The signature on the form is merely the start; the true consent is a living agreement, reaffirmed through trust and transparency at every step of the journey.
Investigators are bound by a strict requirement to conduct the trial in exact compliance with the protocol—a detailed plan they personally sign, formally committing themselves to its procedures. This plan has been approved by ethics committees and regulatory authorities. Adhering to the protocol ensures scientific rigor and consistency. However, there is one profound exception to this rule, revealing the ultimate ethical hierarchy in clinical research.
An investigator can, and in fact must, deviate from the protocol without delay if it is necessary to eliminate an “immediate hazard” to a trial participant. This duty to protect is absolute and supersedes the obligation to follow the study plan. The gravity of this responsibility is captured in GCP guidance:
When an immediate hazard is detected, it MUST be acted on immediately in order to protect participants’ safety and wellbeing.
This is not an action taken lightly. After taking the necessary steps to protect the participant, the investigator must immediately report the deviation and the reasons for it to the trial sponsor, the ethics committee (IRB/IEC), and the relevant regulatory authorities. This powerful exception demonstrates that while generating reliable data is crucial, the investigator’s foremost and non-negotiable priority is the safety and well-being of the human beings in their care.
The role of a clinical trial investigator is far more complex and human-centric than often perceived. They are not only scientists but also site managers overseeing complex logistics, ethical counselors engaging in the deep process of informed consent, and above all, guardians with the ultimate duty to protect participant safety, even if it means breaking from the scientific plan.
The complex web of rules in clinical research isn’t just about good data; it’s about honoring the trust of those who participate. The next time you hear of a medical breakthrough, remember the investigator on the front lines. Their work shows us that the most rigorous science is not a cold, detached process, but a deeply human one, built on a framework where the duty of care does not simply balance the pursuit of knowledge—it enables it.
References
For those interested in gaining our Transcelerate Biopharma-certified courses, please enroll in our ICH GCP E6 R3 courses at https://www.whitehalltraining.com/
#GCPE6R3 #ClinicalTrials #ICHGuidelines #ClinicalResearch #ICH #E6R3 #GCP #WhitehallTraining #CRO #GoodCllinicalPractice #ClinicalTrials
Guidance To Explore
For those wanting to dive deeper into the details:
We’re pleased to share that our ICH GCP E6(R3) courses are now available in multiple languages — ensuring that investigators, coordinators, sponsors, and site teams worldwide can train with clarity and confidence.
💬 Newly available translations:
Spanish
French
German
Italian
Polish
Portuguese
Chinese (Simplified)
Japanese
Korean
These translations support global teams in meeting GCP compliance standards without language barriers — while maintaining the same rigour, accreditation, and assessment integrity as our English version.
🔗 Explore the full GCP E6(R3) course catalogue:
https://whitehalltraining.com/good-clinical-practice-courses
As the industry moves toward the principles-driven approach of R3, it’s essential that every member of the clinical research ecosystem understands not just the requirements — but the reasoning behind them.
Accessible training helps make that possible.
If you’re part of a clinical research site team, this one’s for you.
Our newly updated ICH GCP E6(R3) for Site Investigators course is designed to help site staff confidently apply Good Clinical Practice in everyday trial operations — with a strong focus on practical compliance.
🔍 What you’ll learn:
✔ Investigator responsibilities
✔ Informed consent
✔ Subject safety & data integrity
✔ Delegation & documentation
✔ Essential documents & safety reporting
🎓 Fully compliant & certified
Learners receive CPD-accredited certification (6 CPD points) from the Faculty of Pharmaceutical Medicine, with a unique certificate ID and randomised exam questions for true knowledge validation.
💡 Why this course?
✓ Affordable alternative to costly face-to-face training
✓ Time-saving online modules
✓ Simple bulk licence allocation for teams
✓ Group discount: 10% off for 5+ licences
🛒 Course link:
ICH GCP E6 R3 for Site Investigators → https://www.whitehalltraining.com/good-clinical-practice/site-investigators-r3-version
Give your site teams the confidence and compliance they need to deliver high-quality clinical trials.
Clinical research standards are evolving — and so should your knowledge.
Whitehall Training is proud to introduce the ICH GCP (E6 R3) Refresher Course, designed to help you stay compliant, confident, and current with the latest Good Clinical Practice (GCP) updates.
📘 What’s inside:
This concise, 2-hour refresher provides a focused overview of the ICH E6(R3) revision — the most significant update to GCP in nearly a decade.
You’ll explore:
✅ The new risk-based quality management approach
✅ Enhanced participant protection and ethical standards
✅ Digital and decentralized trial guidance
✅ Updated data governance and integrity principles
🎓 Perfect for professionals who’ve already completed GCP training and need to refresh their certification with the latest regulatory and operational insights.
⏱️ Duration: 2 hours 🏅 CPD Points: 2
👉 Stay ahead in GCP. Enrol today and keep your compliance knowledge future-ready.
![How to Get an ICH E6(R2,R3) Certification [2025]](https://wp.whitehalltraining.com/wp-content/uploads/2025/11/68b6be6eb3569e524962630f_good-clinical-practice-11-scaled.jpg)
Over 80% of clinical trials fail to meet recruitment timelines, often due to non-compliance with Good Clinical Practice (GCP) in researcher guidelines.
In today’s complex clinical research landscape, obtaining ICH E6(R2,R3) certification is paramount for ensuring compliance with international standards and significantly improving study success rates. This certification not only enhances the quality and integrity of clinical trials but also plays a crucial role in safeguarding the rights, safety, and well-being of trial participants.
This blog will guide you through the comprehensive process of achieving ICH E6(R2,R3) certification, providing you with essential knowledge to navigate the intricacies of Good Clinical Practice and elevate your research standards. Whether you’re an experienced professional or new to the field, this information will prove invaluable in advancing your career in clinical research and maintaining rigorous regulatory compliance.
Key takeaways:
ICH E6(R2,R3) certification demonstrates compliance with the Good Clinical Practice (GCP) guidelines established by the International Council for Harmonisation. This internationally recognised standard ensures clinical trials are conducted ethically and scientifically, prioritising participant safety and data credibility.
In clinical research, ICH E6(R2,R3) certification is crucial. It provides a unified standard for the European Union, Japan, and the United States, facilitating mutual acceptance of clinical data by regulatory authorities. This certification showcases commitment to the highest quality and ethical standards in clinical trials.
The ICH E6(R2,R3) history dates back to 1996 when the original guideline was first published. Since then, it has evolved to address the increasing complexity of modern clinical trials. The lateset R3 revision focuses on:
One significant update in ICH E6(R3) is the emphasis on Risk-Based Monitoring. This approach allows for a more flexible and targeted monitoring strategy based on the specific risk level associated with a trial, improving efficiency without compromising quality.
Key benefits of obtaining ICH E6(R2,R3) certification include:
Bottom line: by aligning with the World Medical Association’s Declaration of Helsinki, thereby maximising subject rights, safety, privacy, and well-being. This certification is an essential benchmark for quality and integrity in clinical research, fostering trust among stakeholders and advancing global health initiatives.
We offer comprehensive Good Clinical Practice (GCP) course to help you achieve certification and excel in your clinical research endeavours.
Understanding the ICH E6(R2,R3) guideline is crucial for obtaining certification. Let’s break down the key aspects:
Overview of the ICH E6(R2,R3) document structure:
The guideline is organised into sections covering GCP principles, stakeholder responsibilities, and essential trial documents. It provides a comprehensive framework for ensuring ethical conduct and scientific integrity in clinical research.
Key changes and additions in the R2 revision:
The R2 revision, adopted in 2016, introduced significant updates:
Key Changes and Additions in the R3 Revision (2025):
The R3 revision introduces several important updates and enhancements to strengthen the Good Clinical Practice (GCP) framework:
Focus on risk-based approaches and quality management:
A central theme is the implementation of Risk-Based Quality Management (RBQM). This approach encourages sponsors and investigators to identify, assess, and mitigate risks throughout the clinical trial process, ensuring more efficient and effective trial management.
Resources for accessing and studying the guideline:
Access the full ICH E6(R2,R3) guideline on the official ICH website. Regulatory bodies like the FDA and EMA provide additional guidance documents and training materials to aid in understanding and implementation.
Tips for effective comprehension and interpretation:
Bottom line: A thorough understanding of the ICH E6(R2,R3) guideline is the foundation for developing compliant clinical trial practices and ensuring the protection of human subjects while maintaining data integrity. It’s essential for successful certification and implementation.
To obtain ICH E6(R2,R3) certification, comprehensive training and education are essential. Let’s explore your options for mastering this crucial guideline:
Training Programs:
Introductory courses for newcomers to clinical research
Advanced workshops for experienced professionals
Specialised programmes focusing on specific ICH E6(R2,R3) aspects
Certification preparation courses
Reputable Providers:
Organisations like the MRCT Center offer excellent ICH E6(R2,R3) courses. We at Whitehall Training also provide comprehensive ICH E6(R2,R3) training tailored to your needs.
Training Formats:
Online courses: Offer flexibility and self-paced learning
In-person training: Provides hands-on experience and networking opportunities
Hybrid programmes: Combine both online and in-person elements
Key Topics Covered:
Ethical considerations in clinical research
Quality management systems
Risk-based approaches to clinical trials
Essential documents and record-keeping
Sponsor and investigator responsibilities
Safety reporting and monitoring
Continuous Learning:
Stay updated on regulatory changes, industry best practices, emerging technologies in clinical research, and real-world applications of ICH E6(R2,R3) through ongoing education.
Remember, quality training ensures proper interpretation and application of ICH E6(R2,R3) principles in clinical research practices. By investing in your education, you’ll be well-equipped to implement the guideline effectively and maintain compliance in your clinical trials.
Bottom line: Comprehensive training is crucial for successful ICH E6(R2,R3) certification and implementation. Choose a programme that fits your needs and commit to ongoing learning to stay at the forefront of good clinical practice.
Developing a robust compliance strategy is crucial for successful ICH E6(R2,R3) implementation and certification. Let’s break down the key components:
We offer Good Clinical Practice (GCP) course to help you develop a robust compliance strategy. Our expert-led training ensures you’re well-equipped to meet guideline requirements and demonstrate your commitment to high-quality clinical research practices.
Bottom line: A well-structured compliance strategy is essential for successful ICH E6(R2) implementation and certification. By addressing these components thoroughly, you’ll be well-positioned to meet the guideline’s requirements and showcase your dedication to excellence in clinical research.
Thorough documentation is the backbone of ICH E6(R2,R3) compliance. It’s not just about keeping records; it’s about creating a clear, traceable history of your clinical trial processes. Let’s explore the key aspects of documentation and record-keeping for your certification journey.
Key areas requiring documentation include:
Best practices for maintaining accurate and complete records:
To organise and retrieve records efficiently, consider implementing an electronic document management system with features like version control, audit trails, and secure access.
Data integrity and traceability are crucial. Ensure all processes and decisions can be traced back to their origins, creating a clear audit trail. The investigator should ensure the accuracy, completeness, legibility, and timeliness of the data reported to the sponsor in the CRFs and all required reports. Any changes or corrections should be dated, initialled, and explained to maintain data integrity.
Remember to maintain investigator records for at least 2 years after the last approval of a marketing application in an ICH region, or until there are no pending or contemplated marketing applications, or at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product.
Accurate record keeping is emphasised in ICH GCP E6(R2,R3), which stresses the importance of maintaining complete records that are attributable, legible, contemporaneous, original, accurate, and complete.
We understand the challenges of documentation in clinical trials. Our ICH E6(R2,R3) training courses can help you master these essential skills and ensure your compliance with the latest guidelines.
Bottom line: Proper documentation is crucial for demonstrating compliance and passing audits or inspections. By implementing robust documentation practices, you’ll be well-positioned to achieve and maintain ICH E6(R2,R3) certification.
Regular internal audits are a cornerstone of maintaining ICH E6(R2,R3) compliance and preparing for certification. These audits provide invaluable Internal Audit Benefits, offering tailored insights to identify and rectify process flaws while proactively enhancing operational efficiency. By conducting thorough self-assessments, organisations can ensure they’re meeting the stringent requirements of ICH E6(R2,R3) and be better prepared for external inspections.
To maximise the effectiveness of your internal audit process, develop a comprehensive audit plan and schedule. This should outline the frequency of audits, areas to be covered, and responsible personnel. Aim to audit all critical aspects of your clinical trial processes at least annually, with more frequent checks for high-risk areas.
When conducting internal audits, focus on key areas that align with ICH E6(R2,R3) principles:
As you identify non-conformities during audits, address them promptly and implement corrective actions. This proactive approach not only ensures ongoing compliance but also demonstrates a commitment to continuous improvement – a key aspect of ICH E6(R2).
Preparing for regulatory inspections is the final crucial step. Use the insights gained from internal audits to refine your processes and documentation. Conduct mock inspections to familiarise your team with the inspection process and identify any remaining gaps in your compliance efforts.
We offer comprehensive ICH E6(R2,R3) training courses to help you master these audit and inspection preparation techniques. Our expert-led programmes ensure you’re well-equipped to maintain compliance and achieve certification.
Bottom line: Regular audits and inspection readiness are essential for maintaining ICH E6(R2,R3) compliance and increasing your chances of successful certification. By implementing a robust internal audit program and preparing diligently for inspections, you’ll be well-positioned to demonstrate your commitment to quality and compliance in clinical research.
Selecting the right certification body is crucial for obtaining ICH E6(R2,R3) certification. When researching accredited bodies, look for organisations with a strong reputation in clinical research and recognition by regulatory authorities.
Consider these factors when choosing:
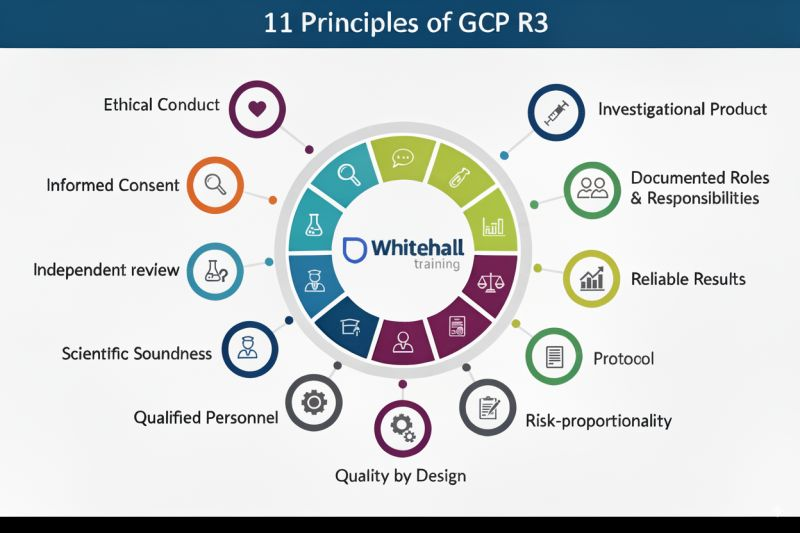
The application process typically involves submitting a formal request with detailed information about your organisation, clinical trial processes, and quality management system. Be prepared to demonstrate how you’ve implemented the 11 principles of good clinical practice outlined in the ICH E6(R2,R3) guideline.
Required documentation and evidence may include:
Timeframes can vary, generally taking several months from application to certification. This process may include initial assessment, documentation review, on-site audits, and corrective actions if necessary. Costs also vary, so request detailed quotes from multiple bodies to ensure you’re getting the best value for your investment.
We offer comprehensive Good Clinical Practice (GCP) course to help prepare you for certification. Our expert-led programmes cover all aspects of GCP, ensuring you’re well-equipped for the application process and ongoing compliance.
Remember, proper preparation is key to successful certification. It demonstrates your commitment to ICH E6(R2) Compliance and ethical and scientific quality standards in clinical trials, enhancing your credibility in the field.
Bottom line: Selecting the right certification body and thoroughly preparing for the application process is crucial for obtaining ICH E6(R2,R3) certification. This step ensures that your organisation meets international standards for clinical trials, protecting subject rights and data credibility while enhancing your competitiveness in the industry.
Obtaining ICH E6(R2,R3) certification is not a one-time achievement but an ongoing commitment to excellence in clinical research. The ICH E6(R2) Compliance guideline emphasises the importance of ongoing compliance and improvement to ensure the safety of research participants and the integrity of data. This final step in your certification journey focuses on implementing processes that foster continuous improvement and maintain long-term compliance.
To stay updated on ICH E6(R2,R3) developments:
These resources provide valuable insights into upcoming changes and best practices in the field.
Implementing feedback mechanisms and performance metrics is crucial for identifying areas of improvement:
Regular review and update of clinical trial processes is essential to ensure ongoing compliance:
Fostering a culture of quality and compliance is perhaps the most critical aspect of continuous improvement:
Bottom line: Continuous improvement ensures long-term compliance and maintains the value of ICH E6(R2,R3) certification. By implementing these strategies, you not only safeguard your certification but also contribute to the advancement of ethical and efficient clinical research practices, ultimately leading to safer and more effective clinical trials.
Obtaining ICH E6(R2,R3)Certification is a crucial step for clinical research professionals, ensuring compliance with international ethical and scientific quality standards. Let’s recap the key steps to achieve this certification:
Remember, compliance with ICH E6(R2,R3) is an ongoing journey. Regularly review and update your practices to maintain certification and stay aligned with evolving industry standards.
The benefits of ICH E6(R2,R3) certification are substantial:
If you haven’t started your certification journey yet, now is the time to begin. The path to excellence in clinical research starts with a strong foundation in Good Clinical Practice. Good Clinical Practice (GCP) course provides comprehensive training to guide you through the process.
For further learning and support, consider exploring these valuable resources:
To conclude, ICH E6(R2) is pivotal in advancing clinical research quality and integrity. By pursuing this certification, you’re not just fulfilling a requirement – you’re contributing to the advancement of medical science and the protection of human subjects. Find out how to get your Good Clinical Practice (GCP) certification today.
![Good Clinical Data Management Practice Guidelines [2025]](https://wp.whitehalltraining.com/wp-content/uploads/2025/11/68b6be6eb3569e5249626330_research-ethics_29-scaled.jpg)
Understanding and implementing these guidelines is essential for researchers, data managers, and clinical trial professionals to maintain data integrity, comply with regulations, and ultimately contribute to the advancement of medical knowledge and patient care.
In this comprehensive guide to GCDMP guidelines for 2024, we’ll provide valuable insights into best practices, regulatory requirements, and emerging trends in clinical data management. You’ll gain practical knowledge on how to implement these guidelines effectively, ensuring your clinical trials meet the highest standards of data quality and integrity.
Key takeaways:
Good Clinical Data Management Practice (GCDMP) guidelines are essential standards ensuring accuracy, consistency, and reliability in clinical trial data. These guidelines form the backbone of credible research outcomes by providing a comprehensive framework for managing clinical data throughout a study’s lifecycle.
The GCDMP definition encompasses the current industry standard for clinical data management, combining best business practices and acceptable regulatory standards. These guidelines are crucial for maintaining data integrity and quality in clinical trials, directly impacting research success and the development of life-saving treatments.
The importance of GCDMP in clinical trials and research is significant:
Without robust data management practices, data error rates in clinical trials can range from 20% to 60%, potentially compromising research validity.
The evolution of GCDMP has been driven by the increasing complexity of clinical trials, the need for standardised practices, technological advancements, and changing regulatory requirements. This evolution ensures that the guidelines remain relevant and effective in the modern research landscape.
The Society for Clinical Data Management (SCDM) plays a pivotal role in establishing and maintaining these guidelines. The SCDM Guidelines provide comprehensive best practices covering essential data management domains, including:
These guidelines are continuously updated by subject matter experts to reflect the latest industry standards and regulatory requirements.
Key objectives of GCDMP include:
GCDMP guidelines are essential standards that ensure the accuracy, consistency, and reliability of clinical trial data, forming the backbone of credible research outcomes. By adhering to these guidelines, researchers can significantly improve data quality, reduce errors, and ultimately contribute to the advancement of medical knowledge and patient care.
Good Clinical Practice jobs often revolve around the core principles of Good Clinical Data Management Practice (GCDMP). These principles are built on several foundational elements that ensure the integrity, accuracy, and reliability of clinical trial data. These core principles are essential for maintaining high standards in clinical research and meeting regulatory requirements.
necessary for reliable study outcomes and regulatory acceptance. Our commitment to these principles underpins our approach to clinical data management, providing a solid framework for conducting high-quality clinical research.
A robust Data Management Plan is the cornerstone of successful clinical research, ensuring data integrity and quality throughout the study lifecycle. This strategic blueprint guides every aspect of data handling, from collection to analysis, and is crucial for maintaining consistency and quality in clinical trials.
Key components of a comprehensive data management plan include:
Tailoring the plan to specific study needs is essential. For instance, a multi-site global trial might require additional focus on data standardisation and cross-cultural validation, while a study involving sensitive patient information may need enhanced security protocols.
Regular review and updates are crucial to keep the plan relevant. We recommend:
Implementing FAIR (Findable, Accessible, Interoperable, Reusable) Data Management principles enhances the plan’s effectiveness and future-proofs your data. This approach not only improves current study quality but also facilitates potential meta-analyses or follow-up studies.
A well-structured data management plan is crucial for maintaining consistency and quality throughout the clinical trial process. It serves as a living document that guides all data-related activities, ensuring that the valuable information collected during the trial is managed effectively, securely, and in compliance with all relevant standards and regulations.
In clinical trials, accurate data collection and rigorous validation are the cornerstone of reliable results. This crucial phase sets the foundation for the entire study, emphasising the need for standardised methods and advanced technologies to maintain data integrity.
Standardised data collection methods, particularly electronic Case Report Forms (eCRFs), have become essential in modern clinical trials. These digital forms ensure consistency across study sites and minimise transcription errors associated with paper-based systems. The implementation of Electronic Data Capture (EDC) systems has revolutionised clinical data management, offering a streamlined approach to capturing patient information while promoting accuracy through built-in features.
Data validation techniques are critical for maintaining data quality and reliability:
Data validation in clinical trials is fundamental as it ensures that the collected data is reliable, accurate, and suitable for analysis.
Managing and resolving data queries promptly is crucial for maintaining study integrity and timelines. A systematic approach to query resolution, involving all relevant stakeholders, ensures timely and accurate responses.
Real-time data cleaning is another vital aspect of the process. This ongoing practice allows for immediate identification and correction of errors, reducing the accumulation of issues that could compromise study outcomes. Implementing automated data cleaning algorithms can significantly improve efficiency and reduce human error.
Rigorous data collection and validation processes are vital for maintaining data integrity and reducing errors in clinical trials. By implementing standardised methods, leveraging EDC systems, and employing comprehensive validation techniques, researchers can ensure the accuracy and reliability of clinical data from the very start. Investing in robust data management practices pays dividends in the form of high-quality, trustworthy research outcomes.
In clinical data management, safeguarding sensitive information is paramount. Robust security measures ensure data integrity throughout the clinical trial process. Let’s explore key aspects of data security and integrity in Good Clinical Data Management Practice (GCDMP).
Protecting data from unauthorised access involves implementing:
Security audits should include thorough examination of security policies and rigorous testing of enforcement mechanisms to ensure comprehensive protection.
Encryption and secure data environments form the backbone of data protection:
Regular data backups and disaster recovery plans are essential for business continuity and data preservation:
Maintaining audit trails and version control is crucial for tracking changes and ensuring data integrity:
Compliance with data protection regulations, such as GDPR and HIPAA, is non-negotiable in clinical data management. These regulations set standards for data privacy and security, and adherence is crucial for maintaining ethical and legal integrity in clinical trials.
It’s important to note that while technological solutions are vital, human factors play a significant role in data security. Human error accounts for 95% of cybersecurity data breaches, underscoring the importance of comprehensive staff training and awareness programmes as part of a holistic security strategy.
Robust security measures are essential to maintain data integrity and protect sensitive patient information throughout the clinical trial process. Implementing these strategies significantly reduces data breach risks and ensures the reliability of research outcomes.
In the rapidly evolving landscape of clinical research, ongoing training for clinical data management (CDM) teams is paramount. As the field continues to advance, it’s crucial for data management professionals to stay abreast of the latest developments, technologies, and regulatory changes. Ongoing Training for CDM Teams ensures that clinical data management teams remain updated on cutting-edge practices and industry standards.
Key areas of focus for training and education include:
Certification programs and continuous education opportunities play a vital role in professional development. Organisations should encourage and support their staff in pursuing relevant certifications, such as the Society for Clinical Data Management’s Certified Clinical Data Manager programme. Attending industry conferences, webinars, and participating in online courses further contribute to the team’s expertise.
Building a culture of quality and compliance is equally important. This involves fostering an environment where team members are encouraged to ask questions, share knowledge, and continuously improve their skills. Regular internal workshops, knowledge-sharing sessions, and mentoring programs for junior team members can contribute to this culture of continuous learning and improvement.
GCDMP and Regulatory Compliance training is particularly crucial for CDM practitioners to ensure high-quality data and adherence to regulatory standards. By focusing on these areas, teams can effectively implement Good Clinical Data Management Practices and adapt to evolving industry standards.
Well-trained and knowledgeable data management teams are crucial for implementing GCDMP effectively and adapting to evolving industry standards. By investing in ongoing education and fostering a culture of continuous improvement, organisations can ensure that their clinical data management processes remain robust, compliant, and at the forefront of industry best practices.
Regulatory compliance is the cornerstone of clinical data management, ensuring the integrity and reliability of trial data. Let’s explore the key standards shaping global clinical research practices.
Good Clinical Practice (GCP) forms the ethical and scientific foundation for clinical trials. These GCP Standards safeguard trial subject rights and ensure data quality, originating from the Declaration of Helsinki.
The International Council for Harmonisation (ICH) plays a pivotal role in setting global standards. The latest ICH E6(R3) guidelines emphasise risk-based quality management in clinical trials, refining previous versions to enhance trial efficiency and data reliability.
Key regulatory bodies like the FDA and EMA enforce specific data management requirements:
These regulations ensure data meets the quality standards necessary for regulatory submissions.
Internationally, ICH GCP Guidelines provide a unified standard for the EU, Japan, and the US. This harmonisation facilitates mutual acceptance of clinical data, crucial for global trials spanning multiple jurisdictions.
Preparing for regulatory inspections involves:
Adhering to regulatory standards is non-negotiable in clinical trials, ensuring that data meets the required quality for submission and approval. By following these global standards, we not only validate our research but also contribute to advancing medical science while protecting trial participants’ rights and well-being.
Implementing best practices and leveraging cutting-edge tools are crucial for optimizing clinical data management processes. These strategies not only enhance efficiency but also significantly improve the quality of data collected and analysed in clinical trials.
Standardised data formats are the cornerstone of efficient clinical data management. The CDISC Standards for Clinical Research, including SDTM (Study Data Tabulation Model) and ADaM (Analysis Data Model), play a vital role in supporting the acquisition, exchange, submission, and archival of clinical research data and metadata. These standards ensure consistency across different studies, facilitate easier data integration and analysis, and support data aggregation and warehousing.
Leveraging technology is another crucial aspect of optimising data management processes:
Implementing risk-based monitoring approaches is becoming increasingly important in clinical trials. This strategy involves focusing monitoring activities on the areas of highest risk to patient safety and data integrity, thereby optimising resource allocation and improving overall trial quality.
Quality assurance and quality control measures are essential components of good clinical data management practice. These include:
These measures ensure consistency and accuracy throughout the data lifecycle.
The benefits of adopting these best practices are significant. Data Management in Clinical Research is a critical phase that leads to the generation of high-quality, reliable, and statistically sound data. By implementing standardised formats like SDTM, clinical researchers can foster data mining and reuse, facilitate sharing, and improve the regulatory review and approval process.
Adopting industry best practices and leveraging cutting-edge tools can significantly enhance the efficiency and quality of clinical data management. By implementing standardised formats, utilising advanced technologies, and focusing on quality assurance, researchers can ensure the integrity and reliability of their clinical trial data, ultimately contributing to more robust and credible research outcomes.
As we look towards the future of clinical data management, exciting innovations are reshaping our field. Let’s explore the trends that will impact how we handle and analyse clinical data in the coming years.
Emerging technologies are leading this transformation:
We’re also seeing a shift towards integrating real-world data and evidence. This approach bridges the gap between controlled clinical environments and real-world scenarios, providing a more comprehensive understanding of patient outcomes.
Interoperability standards are becoming crucial:
Managing big data in clinical trials presents both challenges and opportunities. While the volume can be overwhelming, it offers potential for more nuanced analyses. We’re developing advanced analytics tools and cloud computing solutions to handle these large datasets effectively.
Patient-centred data collection methods are gaining traction. By actively involving patients in the data collection process, we’re improving data quality and patient engagement, leading to more representative and reliable study outcomes.
Staying informed about these trends is crucial for adapting our Good Clinical Data Management Practices (GCDMP) to meet evolving industry needs and technological advancements. We must remain agile and open to new methodologies that enhance the efficiency, accuracy, and value of our clinical data management processes.
Good Clinical Data Management Practice (GCDMP) guidelines are the cornerstone of high-quality clinical research. Let’s recap their critical role in advancing medical science:
Key benefits for stakeholders:
Implementing GCDMP in your organisation:
Implementing GCDMP Guidelines requires commitment, but the benefits far outweigh the costs. By following these steps, you’ll significantly improve your data management practices and contribute to higher-quality clinical research.
Stay up-to-date on GCDMP with these resources:
The GCDMP Best Practices provide a robust framework for ensuring data quality, integrity, and regulatory compliance in clinical trials. By embracing these guidelines, we can continue to advance medical knowledge, develop life-saving treatments, and ultimately improve patient outcomes.
As we look towards the future of clinical research, the role of GCDMP will only grow in importance. The ongoing evolution of technologies like artificial intelligence and machine learning in data management, coupled with the increasing integration of real-world data, underscores the need for adaptable and comprehensive guidelines. GCDMP will continue to serve as a cornerstone for high-quality, reliable, and impactful clinical studies, driving innovation and ensuring patient safety in the ever-changing landscape of medical research.

When we think of a clinical trial sponsor, the image that often comes to mind is that of a well-funded organization—a pharmaceutical company or university—that provides the financial backing for groundbreaking medical research. They are, in the common view, the entity that pays the bills, hoping for a successful outcome that leads to a new treatment or a prestigious publication.
But when a trial faces challenges—from unexpected safety events to questions of data integrity—who is ultimately accountable? Who is responsible for protecting the rights and safety of the human participants who volunteer for these trials? The answer, as defined by the global standard for research ethics and quality known as Good Clinical Practice (GCP), places a profound and often surprising weight squarely on the sponsor’s shoulders.
This article reveals four of the most impactful responsibilities that clinical trial sponsors must uphold. Going far beyond simply funding the research, these duties position the sponsor as the ultimate guardian of a trial’s ethical conduct and scientific integrity.
In the complex world of clinical research, sponsors frequently delegate day-to-day trial management tasks to specialized companies known as Contract Research Organizations (CROs) or other service providers. These organizations may handle everything from site monitoring to data management, creating a network of partners to execute the study.
Despite this delegation, GCP guidelines are uncompromising on one critical point: the sponsor retains full and ultimate responsibility for every aspect of the trial. They cannot outsource their accountability. This means the sponsor is answerable for the quality of the trial, the integrity of the data, and, most importantly, the protection of participants’ rights, safety, and well-being. This principle is essential for preventing a diffusion of accountability, ensuring that a single, identifiable entity can be held responsible for the entire trial from start to finish. This accountability is so absolute that it extends even to work that their primary CRO may subcontract to other vendors, ensuring there are no gaps where responsibility could be lost.
“The sponsor retains ultimate responsibility for sponsor’s trial-related activities, including protection of participants’ rights, safety and well-being and reliability of the trial data.”
A common misconception about clinical trial regulations is that they are a rigid, “one-size-fits-all” checklist that must be applied with equal force to every study, regardless of its size or complexity. However, modern GCP principles have moved away from this inflexible model.
The sponsor’s responsibility is not merely to enforce a long list of rules but to implement a “proportionate and risk-based approach” to quality management. This concept, known as “Quality by Design (QbD),” requires the sponsor to proactively identify the factors that are “critical to quality”—that is, the data and processes most essential for protecting participant safety and ensuring reliable results. The sponsor must then focus their resources and oversight on managing the highest risks to these critical factors. This shift is not just about efficiency; it represents an ethical imperative. It moves the field away from a “compliance for compliance’s sake” mindset toward a model that intelligently focuses resources on what matters most: protecting participants from genuine risks and ensuring the scientific question can be answered reliably, thereby honoring the contribution of every volunteer.
“Sponsors should focus on trial activities essential to ensuring the protection of human subjects and the reliability of the trial results. Quality management includes the efficient design of clinical trial protocols, data collection tool and procedures, and the collection of information that is essential to decision making.”
One of the most fundamental duties of a sponsor is to select investigators who are qualified, experienced, and ethical. However, this responsibility does not end once the contracts are signed. The sponsor must actively monitor the investigator’s work throughout the trial to ensure they are complying with the protocol and to detect any potential issues, including misconduct or fraud.
A powerful example of what can go wrong is the “KETEK Case,” an antibiotic trial where an investigator enrolled over 400 participants in just five months. An inspection by the U.S. Food and Drug Administration (FDA) uncovered widespread fraud, including the “complete fabrication of participant enrolment.” The investigator ultimately received a 57-month prison sentence. Crucially, a warning letter was also issued to the trial’s sponsor for critical failures in their oversight duties. These failures included:
This responsibility transforms the sponsor-investigator relationship from one of simple funding to one requiring active, ongoing verification. It establishes an essential “trust but verify” framework, making the sponsor an active guardian of scientific integrity to protect both patients and the validity of the research itself.
It seems intuitive that the organization paying for a clinical trial would have exclusive ownership and control over the data it generates. However, GCP guidelines establish a crucial check and balance that runs counter to this assumption.
The sponsor must ensure that the investigator—the physician or researcher with direct medical responsibility for the participants at a clinical site—retains access to and control over the data collected at that site. The sponsor cannot have sole or exclusive control of the data acquisition tools. This shared control is not merely a technicality; it is a critical safety feature. It empowers the investigator—the one with direct medical responsibility—to access real-time data to make crucial decisions about a participant’s eligibility, ongoing treatment, and continued safe participation in the trial. This prevents data from being altered or misinterpreted without the knowledge of the person closest to the patient.
“The sponsor should not have exclusive control of data captured in data acquisition tools and should ensure that the investigator has access to the required data for retention purposes and that the investigator receives instructions on how to navigate systems, data and relevant metadata for the trial participants under their responsibility.”
The role of a clinical trial sponsor extends far beyond the financial. As these four responsibilities illustrate, the sponsor is the central pillar of accountability, tasked with ensuring a study is not only scientifically sound but ethically unimpeachable. They carry the burden of ultimate responsibility, which cannot be delegated; they must proactively design quality into a trial, not just police a checklist; they serve as a critical line of defense against fraud; and they must share control of the trial’s precious data with the investigators on the front lines.
Ultimately, the sponsor acts as the conscience of the clinical trial. Their role is a profound stewardship rooted in ethical oversight, proactive management, and unwavering accountability for every person who contributes to the research. As medical research relies more on global collaboration and complex data systems, how can we ensure this profound sense of stewardship remains at the very heart of innovation?
References
For those interested in gaining our Transcelerate Biopharma-certified courses, please enroll in our ICH GCP E6 R3 courses at https://www.whitehalltraining.com/
#GCPE6R3 #ClinicalTrials #ICHGuidelines #ClinicalResearch #ICH #E6R3 #GCP #WhitehallTraining #CRO #GoodCllinicalPractice #ClinicalTrials
Guidance To Explore
For those wanting to dive deeper into the details: